Mitochondrial disorders represent a diverse group of conditions with a primary defect in electron transport chain function. Although other conditions, such as amino acid and fatty acid oxidation defects, also occur in the mitochondria, they are not traditionally considered part of the mitochondrial disorders. Many of the mitochondrial myopathies were originally characterized by acronyms based upon the phenotypic presentation. For example, MELAS refers to Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes. The era of mitochondrial medicine expanded rapidly after the identification of a point mutation responsible for Leber’s hereditary optic neuropathy (LHON, G11778A), chronic progressive external ophthalmoplegia (CPEO, mitochondrial DNA deletions) and MELAS (A3243G). Since that time there has been a massive proliferation of the number of point mutations in the mitochondrial genome ascribed to phenotypic characteristics.
In addition to the vast array of mutations found within the maternally inherited mitochondrial DNA (mtDNA), there is an increasing recognition of the mitochondrial disorders arising from mutations within the nuclear genome and following Mendelian genetic inheritance patterns. For example, the mutations responsible for a number of autosomal recessive conditions have been found, including myo-neuro-gastrointestinal encephalomyopathy (MNGIE, thymidine phosphorylase), complex I (NDUF) and IV (SURF-1) Leigh’s disease, mtDNA depletion (dGuOK, TK) and some forms of mtDNA deletion syndromes (polymerase gamma, twinkle, ANT). Autosomal dominant inheritance patterns have also been seen in rare cases of mtDNA deletion syndromes.
Irrespective of the mutation, many of the cellular consequences of mitochondrial dysfunction can be linked to a decrease in aerobic energy production and/or an increased production of free radicals. Given that the mitochondrion is the final common pathway for the oxidative decarboxylation of fats, proteins, and carbohydrates, it is understandable that mitochondrial dysfunction can impair cellular energy metabolism, which can be particularly apparent during periods of superimposed metabolic stress (exercise, infection, prolonged fasting). Given the ubiquitous presence of mitochondria in all tissues except red blood cells, there is often widespread tissue involvement (mitochondrial cytopathies); however, most patients will have varying degrees of muscle symptoms (decreased endurance or weakness), and these forms can be termed ‘mitochondrial myopathies.’ Recent reviews of the mitochondrial cytopathies can be found in Tarnopolsky and Raha (2005) and Dimauro et al. (2006).
Fatty Acid Oxidation Defects
FAODs ultimately impair b-oxidation of lipid within the mitochondrial matrix. The main defects that have been identified include transport of long-chain fat across the mitochondrial membrane (i.e., CPT deficiency); transport of carnitine into the cell (i.e., carnitine transporter deficiency); and the majority of defects attributed to mutations in b-oxidation directly (i.e., long-chain acyl-CoA dehydrogenase (LCAD), medium-chain acyl-CoA dehydrogenase (MCAD), and trifunctional protein (TFP) deficiencies). These disorders are inherited with an autosomal recessive inheritance pattern. The more severe variants present in infancy or childhood with a primary liver or encephalopathic picture, while the adult-onset forms are predominantly myopathic. The main FAODs presenting in adulthood include CPT II, TFP, and very-long-chain acyl-CoA dehydrogenase deficiencies. Further reading in this area can be found in the reviews by Tein (1996) and Vockley et al. (2002).
Glycogen Storage Disease
GSD refers to a group of disorders characterized by genetic mutations in glycogen synthesis, glycogenolysis, or glycolysis. The pathology results from an inability to break down glycogen to maintain plasma glucose concentration (e.g., hepatic forms such as hepatic phosphorylase deficiency or glucose-6-phosphatase deficiency), abnormal tissue storage and cirrhosis (e.g., branching enzyme deficiency), or the myopathic forms that inhibit muscle glycogenolysis or glycolysis (e.g., McArdle’s disease, Tarui’s disease, etc.). The hepatic forms usually result in hypoglycemia as the main clinical manifestation, with type IV also leading to cirrhosis due to the accumulation of abnormal nonbranched glycogen molecules. The myopathic forms usually result in muscle cramping and premature fatigue particularly during high-intensity exercise when there is the obligatory use of anaerobic pathways. Some of the myopathic forms of GSD result in fixed muscle weakness such as Pompe’s disease (GSD II), whereas others such as McArdle’s disease (GSD V) may develop a more indolent proximal myopathy later in life. With the exception of phosphorylase b kinase deficiency and phosphoglycerate kinase deficiency, which are X-linked recessive conditions, the remainder of the GSDs are autosomal recessive in their inheritance pattern.
The more common myopathic forms of GSD, in approximate order of frequency, include phosphorylase deficiency (McArdle’s disease, GSD V), acid maltase deficiency (Pompe’s disease, GSD II), phosphorylase b kinase deficiency (GSD VIII), and phosphofructokinase (PFK) deficiency (Tarui’s disease, GSD VII). Less common myopathic forms include debranching enzyme (GSD III), phosphoglycerate kinase (GSD IX), phosphoglycerate mutase (GSD X), and lactate dehydrogenase (GSD XI) deficiencies.
Clinical Presentation
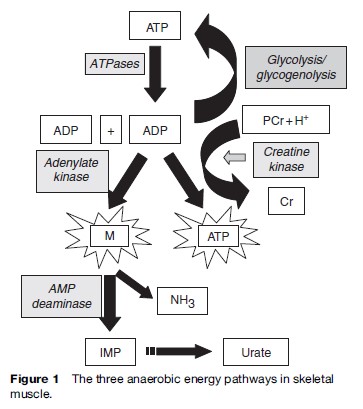
It is important to note that many of the metabolic myopathies present with symptoms during exercise. During higher intensity exercise such as sprinting, or at the onset of aerobic exercise, there is predominantly an anaerobic component, with the adenylate kinase/myoadenylate deaminase pathway being quantitatively the least important, followed by the CK-mediated pathway of phosphocreatine hydrolysis, and anaerobic glycolysis and glycogenolysis (Figure 1).
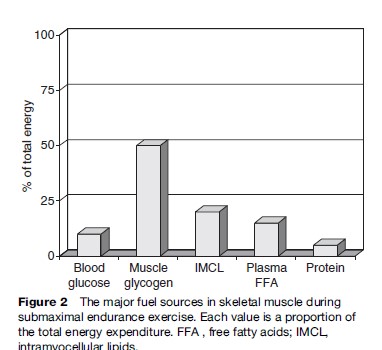
With endurance-type activity, the proportion of oxidized fuels changes as a function of exercise intensity, training status, and gender; however, at moderate-intensity exercise (less than 50% of maximal aerobic capacity (VO2max)), the main sources of fuels are plasma free fatty acids and blood-borne glucose. As the intensity of exercise increases there is proportionately greater utilization of intramuscular glycogen and intramyocellular lipids, with almost exclusive utilization of intramuscular glycogen at intensities closer to a 10-km or marathon race pace. The mitochondria are obligatory for the oxidative metabolism of carbohydrate fat and protein, thus representing the final common pathway of aerobic energy transduction. The main fuel sources during endurance activity at approximately 65% of the VO2max are presented in Figure 2.
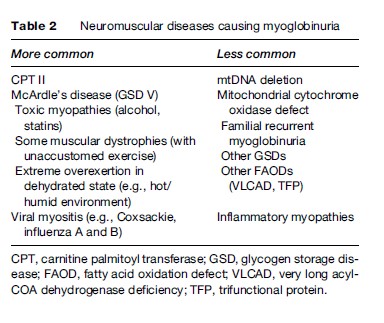
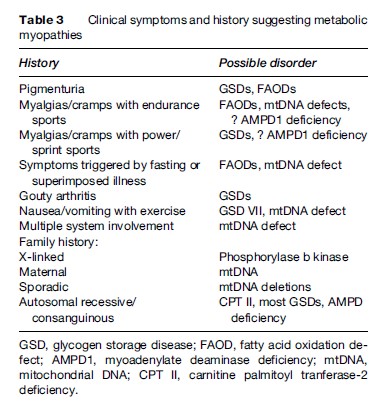
Under normal circumstances, the ATP content of skeletal muscle is tightly regulated and does not decrease; however, with the metabolic myopathies it is possible to reach a state of metabolic crisis and induce rhabdomyolysis, which can lead to myoglobinuria and subsequent renal failure. A list of some of the causes of myoglobinuria is found in Table 2. A list of the common symtoms of the metabolic myopathies is presented in Table 3.
Mitochondrial Disorders
There is extreme phenotypic and genotypic heterogeneity in the mitochondrial cytopathies. For example, patients with the MELAS A3243G gene mutation can present with later adult-onset deafness and diabetes, or in infancy with fatal encephalomyopathy with seizures and stroke-like episodes. Conversely, patients with a wide array of specific point mutations, mtDNA deletions, and nuclear defects can present with encephalopathy, muscle fatigue or ptosis and hearing loss. Due to the impairment in aerobic energy transduction, most patients have very low maximal oxygen uptake (VO2max), which renders daily activities much more stressful and taxing and patients often present with exercise intolerance. Although the exercise intolerance is often overshadowed by more significant neurological symptoms, most adult patients will have quite severe exercise intolerance, often dating back to childhood. These individuals were often labeled as being ‘the worst athlete in the school’ and frequently avoided physical activity. Unlike the FAODs and GSDs, it is rare for patients to have rhabdomyolysis with resultant myoglobinuria; however, this can be seen in cytochrome b, cytochrome c oxidase (COX), and MELAS A3260G mutations.
Figure 2 The major fuel sources in skeletal muscle during submaximal endurance exercise. Each value is a proportion of the total energy expenditure. FFA , free fatty acids; IMCL, intramyocellular lipids. GSD, glycogen storage disease; FAOD, fatty acid oxidation defect; AMPD1, myoadenylate deaminase deficiency; mtDNA, mitochondrial DNA; CPT II, carnitine palmitoyl tranferase-2 deficiency.
In addition to exercise intolerance, patients with mitochondrial myopathies frequently have headache, nausea, and vomiting induced by exercise and can become frankly encephalopathic. We have seen one pedigree with the MELAS A3260G gene mutation where exercise-induced deafness was a characteristic feature. The symptoms of exercise intolerance are much worse when individuals have a superimposed infection or are in the fasted state, which are conditions where a greater reliance on mitochondrial energy function is required.
In addition to exercise intolerance and the extensive list of other clinical features that can accompany these disorders (ptosis, ophthalmoplegia, hearing loss, strokelike episodes, migraine, headaches, cardiomyopathy,ataxia, encephalopathy, etc.), another muscle-related symptom can be fixed weakness. We have had several pedigrees with the MELAS A3243G gene mutation where fixed proximal weakness to the point of respiratory failure was a presenting feature. In most patients, however, strength is reasonably well preserved, and the factors predisposing some patients to severe weakness are currently unclear.
Fatty Acid Oxidation Defects
Many patients with myopathic FAODs are asymptomatic without the superimposition of a metabolic stress. In children, the two main metabolic stressors are superimposed illnesses, such as viral illness, or prolonged fasting, nausea, and vomiting with decreased fluid and caloric intake, where significant weakness, lethargy, and even encephalopathy can occur. The adult myopathic forms of FAOD usually present during endurance type activity, particularly if it is prolonged, performed in the fasted state, or with a superimposed viral illness. These individuals can often perform high-intensity activity for short periods of time without difficulty; however, when the utilization of lipid becomes more important, such as during longer-term endurance activity, they often experience cramps, muscle discomfort, and inability to continue the activity. There is often pigmenturia later in the day or the next day following such an activity, and the muscles remain very sore for several days. In some cases severe myalgias with rhabdomyolysis and renal failure can be the presenting symptom of FAOD.